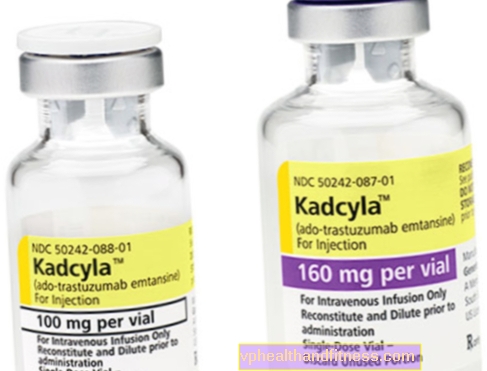
1 frasco para injectáveis contém 100 mg (160 mg) de pó para reconstituição de 5 ml (8 ml) de trastuzumab emtansina 20 mg / ml concentrado para perfusão após reconstituição.
| Nome | Conteúdo da embalagem | A substância ativa | Preço 100% | Última modificação |
| Kadcyla | 1 frasco, pó para preparação solução final para inf. | Trastuzumab emtansina | 2019-04-05 |
Açao
Droga anticâncer. Trastuzumabe emtansina é um conjugado anticorpo-droga que contém trastuzumabe, um anticorpo monoclonal anti-HER2 IgG1 humanizado, covalentemente ligado a DM1 (derivado de maitansina, inibidor de microtúbulo) por meio de uma ligação de tioéter MCC estável (4- ciclohexano-1-carboxilato) . Emtansina é o complexo MCC-DM1. A conjugação do DM1 com o trastuzumabe provoca a ação seletiva de fármacos citotóxicos contra as células tumorais com superexpressão de HER2, aumentando a concentração intracelular de DM1 diretamente nas células tumorais. Após a ligação ao HER2, o trastuzumabe emtansina é internalização mediada por receptor seguida por degradação lisossomal, liberando catabólitos contendo DM1 (principalmente lisina-MCC-DM1). O mecanismo de ação do trastuzumabe emtansina é mediado pela atividade do trastuzumabe e do DM1. O trastuzumabe emtansina, assim como o trastuzumabe, liga-se ao domínio IV do domínio extracelular (ECD) do receptor, bem como aos receptores Fcγ e ao complemento C1q. Além disso, inibe a atividade do domínio ECD do receptor HER2, inibe a sinalização da via da fosfatidilinositol 3-quinase (PI3-K) e medeia a citotoxicidade celular dependente de anticorpos (ADCC) em células de câncer de mama humano que superexpressam HER2. O DM1 se liga à tubulina. Ao inibir a polimerização da tubulina, tanto o DM1 quanto o trastuzumabe emtansina fazem com que as células parem na fase G2 / M do ciclo celular, levando à morte celular por apoptose. O linker MCC reduz a liberação sistêmica de DM1 e aumenta sua concentração no local de destino. O trastuzumabe emtansina é desconjugado e depois catabolizado por proteólise nos lisossomas celulares. O DM1 é metabolizado principalmente pelo CYP3A4 e em menor extensão pelo CYP3A5. O T0,5 do trastuzumab é de aproximadamente 4 dias. Não houve acúmulo de trastuzumabe emtansina após múltiplas infusões intravenosas administradas em um intervalo de 3 semanas. A idade não teve efeito sobre a farmacocinética de trastuzumab entansina.
$config[ads_text1] not found
Dosagem
Por via intravenosa. A preparação deve ser prescrita por um médico e administrada sob a supervisão de profissionais de saúde com experiência no tratamento de pacientes com câncer. Pacientes recebendo trastuzumabe emtansina devem ter câncer HER2-positivo - pontuação de imunohistoquímica (IHC) 3+ ou razão ≥2 na hibridização in situ (ISH). Os testes devem ser realizados usando testes de diagnóstico in vitro (IVD) com marcação CE. Quando o teste CE IVD não estiver disponível, o teste deve ser realizado com outro teste validado. A fim de evitar erros médicos, é importante verificar o rótulo do frasco para injetáveis para se certificar de que o medicamento a ser preparado e administrado é Kadcyla (trastuzumab emtansina) e não Herceptin (trastuzumab). A dose recomendada é de 3,6 mg / kg. administrado por perfusão intravenosa a cada 3 semanas (ciclo de 21 dias). Os pacientes devem ser tratados até a progressão do tumor ou até a obtenção de toxicidade inaceitável. A dose inicial deve ser administrada como uma perfusão intravenosa de 90 minutos. O local da injeção deve ser monitorado cuidadosamente devido à possibilidade de penetração subcutânea do medicamento durante a administração. Se a infusão anterior foi bem tolerada, as doses subsequentes podem ser administradas como uma infusão de 30 minutos. Os pacientes devem ser observados durante a infusão e por pelo menos 30 minutos depois. A taxa de infusão deve ser diminuída ou interrompida se o paciente desenvolver sintomas relacionados à infusão. Trastuzumab emtansina deve ser descontinuado em caso de reações à perfusão com risco de vida. Devem estar disponíveis medicamentos para reações alérgicas / anafiláticas à perfusão, bem como equipamento de emergência para uso imediato. Se uma dose planejada for esquecida, ela deve ser administrada o mais rápido possível; não espere até o próximo ciclo. O esquema de dosagem deve ser ajustado para manter o intervalo de dosagem de 3 semanas. A próxima dose deve ser administrada de acordo com as recomendações de dosagem. O manejo das reações adversas sintomáticas pode incluir a interrupção periódica, redução da dose ou interrupção do tratamento. Esquema de redução da dose (dose inicial 3,6 mg / kg): primeira redução da dose 3 mg / kg; segunda redução da dose 2,4 mg / kg pc; se for necessária uma redução adicional da dose, o tratamento deve ser interrompido. Diretrizes de modificação de dose para elevações de AST e ALT: Grau 2 (> 2,5 a ≤5 x ULN) - nenhum ajuste de dose necessário; grau 3 (> 5 a ≤20 x LSN) - use a preparação se AST / ALT diminuir para o grau ≤ 2 (> 2,5 a 20 x LSN) - interromper o tratamento. Regras de modificação de dose para hiperbilirrubinemia: grau 2 (> 1,5 a ≤3 x LSN) - use a preparação quando o nível de bilirrubina diminuiu para grau ≤1. (> ULN a 1,5 x ULN), nenhum ajuste de dose necessário; Grau 3 (> 3 a ≤10 x LSN) - use a preparação se a concentração de bilirrubina cair para o grau ≤ 1 (> LSN a 1,5 x LSN), então reduza a dose (ver esquema de redução da dose); Grau 4 (> 10 x LSN) - interromper o tratamento. Regras de modificação de dose para trombocitopenia: grau 3 (concentração de plaquetas de 25.000 a 3) - use a preparação quando a concentração de plaquetas for ≤1. (por exemplo, ≥75.000 / mm3); nenhum ajuste de dose necessário; Grau 4 (concentração de plaquetas 3) - use a preparação quando a concentração de plaquetas atingir ≤1. (por exemplo, ≥75.000 / mm3), em seguida, reduza a dose (ver esquema de redução de dose). Princípios de modificação da dose em pacientes com disfunção ventricular: FEVE 45% - continuar a terapia; FEVE 40% a ≤45% com redução simultânea da fração de ejeção por - não use a preparação; Reavalie a FEVE em 3 semanas, se a FEVE ainda for ≥10 pontos percentuais da linha de base, interromper o tratamento; ICC sintomática - interromper o tratamento. Crianças e adolescentes: A segurança e eficácia em crianças e adolescentes com menos de 18 anos de idade não foram estabelecidas, uma vez que o cancro da mama disseminado (MBC) não é conhecido nesta população. Grupos especiais de pacientes. A terapia deve ser temporariamente interrompida em pacientes que desenvolvem neuropatia periférica de Grau 3 ou 4 até atingir Grau ≤2; ao reiniciar o tratamento, pode-se considerar a redução da dose conforme descrito no esquema de redução da dose. Nenhum ajuste de dose é necessário para pacientes ≥ 65 anos de idade; não existem dados suficientes para determinar a segurança e eficácia da terapia em pacientes com idade ≥75 anos. Não é necessário ajuste da dose inicial em pacientes com insuficiência renal leve ou moderada; A necessidade potencial de ajuste da dose em pacientes com insuficiência renal grave não pode ser determinada e, portanto, esses pacientes devem ser monitorados cuidadosamente. Não é necessário ajuste da dose inicial em pacientes com insuficiência hepática leve ou moderada. Trastuzumab emtansina não foi estudado em pacientes com insuficiência hepática grave; deve-se ter cuidado ao tratar pacientes com insuficiência hepática devido à hepatotoxicidade conhecida observada durante o tratamento. Forma de dar. A preparação deve ser dissolvida e diluída pelo pessoal médico e administrada por perfusão intravenosa. Não deve ser administrado em bólus ou injeção rápida.
$config[ads_text2] not foundIndicações
Monoterapia de pacientes adultos com câncer de mama HER2-positivo, inoperável localmente avançado ou metastático, previamente tratados com trastuzumabe e um taxano, em combinação ou separadamente. Pacientes que foram previamente submetidos a tratamento para doença localmente avançada ou generalizada, ou que tiveram recidiva durante ou dentro de 6 meses após completar a terapia adjuvante.
Contra-indicações
Hipersensibilidade à substância ativa ou outros ingredientes da preparação.
Precauções
Casos de doença pulmonar intersticial (DPI), incluindo pneumonia, foram relatados em ensaios clínicos com trastuzumabe emtansina; alguns foram associados a insuficiência respiratória aguda ou foram fatais. Recomenda-se interromper o tratamento com a preparação em pacientes com diagnóstico de DPI ou pneumonia. Pacientes com dispneia em repouso, associada a complicações de câncer avançado e doenças associadas, podem ter maior risco de desenvolver complicações pulmonares. Durante o tratamento com a preparação, foram observados hepatotoxicidade e doenças graves do fígado e do trato biliar, incluindo hipertrofia nodular regenerativa (NRH) do fígado e mortes devido a lesão hepática induzida por drogas; comorbidades e / ou medicamentos concomitantes com potencial hepatotóxico também podem ter sido afetados. A função hepática deve ser monitorada antes do início do tratamento e antes da dosagem subsequente. Pacientes com elevação de ALT basal (por exemplo, associada a metástases hepáticas) estão predispostos ao desenvolvimento de insuficiência hepática e apresentam maior risco de toxicidade hepática de Grau 3-5 ou níveis laboratoriais hepáticos elevados. Os casos de NRH foram diagnosticados por biópsias hepáticas. O desenvolvimento de NRH deve ser considerado em todos os pacientes com sinais clínicos de hipertensão portal e / ou quadro semelhante a cirrose observado na TC do fígado, mas com níveis normais de transaminases séricas e nenhuma outra evidência de cirrose. Se NRH for diagnosticado, o tratamento com a preparação deve ser interrompido. Não foi estudado em pacientes com transaminases séricas> 2,5 x LSN (limite superior do normal) ou com bilirrubina total> 1,5 x LSN antes do início do tratamento. No caso de atividade das transaminases séricas> 3 x LSN e, simultaneamente, concentração total de bilirrubina> 2 x LSN, o tratamento deve ser interrompido. Deve-se ter cuidado ao tratar pacientes com insuficiência hepática. Existe um risco aumentado de disfunção ventricular esquerda durante o tratamento. Em pacientes tratados com trastuzumabe-entansina, observou-se redução da fração de ejeção do ventrículo esquerdo (FEVE) 50 anos, com baixo valor inicial de FEVE (25 kg / m2). Os testes de função cardíaca padrão (ecocardiograma ou angiografia com radioisótopos múltiplos) devem ser realizados antes do início do tratamento e em intervalos regulares (por exemplo, a cada 3 meses). A FEVE dos pacientes no início do estudo foi ≥50% na maioria dos ensaios clínicos. Pacientes com insuficiência cardíaca congestiva, arritmias graves que requerem tratamento, história de infarto do miocárdio ou angina pectoris instável nos 6 meses anteriores à randomização ou com dispneia em repouso devido a doença neoplásica avançada foram excluídos do estudo. No caso de distúrbios do ventrículo esquerdo, a próxima dose deve ser adiada ou a terapia deve ser interrompida. Não foi estudado o efeito do tratamento com trastuzumabe emtansina em pacientes que interromperam o tratamento com trastuzumabe devido a uma reação relacionada à infusão; a terapia com este medicamento não é recomendada neste grupo de pacientes. A terapia deve ser interrompida em pacientes que desenvolverem uma reação grave relacionada à infusão até a resolução dos sintomas. O reinício da terapia deve ser considerado com base no julgamento clínico da gravidade da reação. O tratamento deve ser interrompido no caso de uma reação à perfusão com risco de vida. Não foi estudado o efeito do tratamento com trastuzumabe-entansina em pacientes que interromperam o tratamento com trastuzumabe devido à hipersensibilidade; O uso de trastuzumabe emtansina nesses pacientes não é recomendado. Os pacientes devem ser monitorados quanto a reações de hipersensibilidade / alérgicas; os sintomas podem ser os mesmos que as reações relacionadas à infusão; reações anafiláticas graves foram observadas. Se houver hipersensibilidade (com aumento de reações durante infusões subsequentes), o tratamento com trastuzumabe-entansina deve ser interrompido.Devido ao risco de eventos hemorrágicos (incluindo hemorragia do SNC, respiratória e gastrointestinal), deve-se ter cuidado e considerar-se a monitoração adicional quando coadministrado com anticoagulantes ou medicamentos antiplaquetários. Recomenda-se que a contagem de plaquetas seja monitorada antes de cada dose de trastuzumabe emtansina. Pacientes com trombocitopenia (≤100.000 / mm3) e pacientes recebendo terapia anticoagulante (por exemplo, varfarina, heparina, heparinas de baixo peso molecular) devem ser monitorados de perto durante o tratamento com trastuzumabe emtansina. O trastuzumab emtansina não foi estudado em doentes com contagens de plaquetas ≤100.000 / mm3 antes do início da terapêutica. Em caso de agravamento da trombocitopenia para Grau 3 ou superior (3), a preparação não deve ser usada até que a toxicidade diminua para Grau 1 (≥75.000 / mm3). Em ensaios clínicos com trastuzumab emtansina, ocorreu neuropatia periférica de grau 1, principalmente sensorial. Pacientes com neuropatia periférica de Grau ≥3 foram excluídos da participação em ensaios clínicos. O tratamento de pacientes com neuropatia periférica de grau 3 ou 4 deve ser interrompido periodicamente até que a complicação seja reduzida para grau ≤2. Os pacientes devem ser monitorados regularmente quanto a sinais de neurotoxicidade. Para melhorar a rastreabilidade das preparações biológicas, o nome comercial do medicamento administrado deve ser claramente escrito (ou declarado) no prontuário do paciente. A segurança e eficácia do medicamento em crianças e adolescentes com menos de 18 anos de idade não foram estabelecidas, uma vez que não foi encontrado cancro da mama disseminado nesta população. Não deve ser administrado em bólus ou injeção rápida.
$config[ads_text3] not found
Atividade indesejável
Muito comuns: infecção do trato urinário, trombocitopenia, anemia, hipocalemia, insônia, neuropatia periférica, dor de cabeça, sangramento, epistaxe, tosse, dispneia, estomatite, diarreia, vômito, náusea, constipação, boca seca, dor abdominal , erupção cutânea, dor musculoesquelética, artralgia, mialgia, fadiga, febre, astenia, calafrios, aumento das transaminases. Comum: neutropenia, leucopenia, hipersensibilidade, tontura, disgeusia, comprometimento da memória, síndrome do olho seco, conjuntivite, distúrbios visuais, lacrimejamento aumentado, disfunção ventricular esquerda, hipertensão, dispepsia, sangramento gengival, prurido cutâneo, alopecia, doença das unhas , eritrodisestesia palmo-plantar, urticária, edema periférico, aumento da fosfatase alcalina, reações relacionadas com a perfusão (vermelhidão da pele, arrepios, febre, dispneia, tensão arterial baixa, respiração ruidosa, broncoespasmo, taquicardia). Pouco frequentes: pneumonia (DPI), hepatotoxicidade, insuficiência hepática, hiperplasia nodular regenerativa, hipertensão portal, extravasamento no local da injeção (eritema, sensibilidade, irritação da pele, dor ou inchaço). Além disso, foi observada hiperbilirrubinemia. Em ensaios clínicos, ocorreram aumentos nas transaminases séricas (Grau 1-4) durante o tratamento com trastuzumab emtansina e foram geralmente transitórios. A elevação das transaminases foi na maioria das vezes transitória, com pico no 8º dia após a dosagem. Depois disso, a toxicidade diminuiu para Grau 1 ou foi resolvida antes do próximo ciclo. Houve também um efeito cumulativo (a porcentagem de pacientes com elevação Grau 1-2 ALT / AST aumentou com os ciclos subsequentes). Em pacientes com níveis elevados de transaminase, a maioria dos pacientes experimentou redução da toxicidade de Grau 1 ou recuperação em 30 dias após a última dose de trastuzumabe-entansina. Disfunção ventricular esquerda foi encontrada em 2,2% dos pacientes participantes de ensaios clínicos. Na maioria dos casos, as diminuições da FEVE de grau 1 ou 2 foram assintomáticas. Toxicidade de grau 3 ou 4 foi observada em 0,4% dos pacientes, geralmente durante os ciclos iniciais de tratamento (1–2). Episódios graves de eventos hemorrágicos (Grau ≥3) foram relatados em 2,2% de todos os pacientes. A trombocitopenia ocorreu em 24,9% dos pacientes em ensaios clínicos e foi a reação adversa mais comum que levou à descontinuação da terapia. Em ensaios clínicos, a incidência e gravidade da trombocitopenia foram superiores em doentes asiáticos. Alguns pacientes que desenvolveram essas complicações também foram tratados com terapia anticoagulante. Houve episódios hemorrágicos fatais e complicações hemorrágicas graves, incluindo hemorragia do SNC 5,3% dos doentes com resultados positivos para anticorpos anti-trastuzumab emtansina.
$config[ads_text4] not foundGravidez e lactação
O uso de trastuzumab emtansina em mulheres grávidas não é recomendado. Antes de engravidar, as mulheres devem ser informadas sobre a possibilidade de danos ao feto. No entanto, as pacientes que engravidam devem procurar um médico imediatamente. Se uma mulher grávida for tratada com trastuzumabe emtansina, é recomendado o monitoramento cuidadoso por uma equipe multidisciplinar. Trastuzumab pode causar dano fetal ou morte se administrado a uma mulher grávida. Em crianças de mulheres grávidas tratadas com a preparação, foram relatados casos de oligoidrâmnio, alguns deles levando a hipoplasia pulmonar fatal. O DM1, o componente citotóxico do trastuzumabe emtansina, pode ser teratogênico e potencialmente embriotóxico. As mulheres devem interromper a amamentação antes de iniciar a terapia com trastuzumabe-entansina. Os pacientes podem começar a amamentar 7 meses após a interrupção do tratamento. Mulheres com potencial para engravidar devem usar métodos contraceptivos eficazes durante o tratamento com o medicamento e por 7 meses após a última dose de trastuzumabe-entansina. Métodos eficazes de contracepção também devem ser usados por homens ou suas parceiras.
Comentários
Os doentes que experimentam reações relacionadas com a perfusão não devem conduzir ou utilizar máquinas até que estes sintomas tenham desaparecido. Informações preparadas com base em SPC de 13.07.2017 O SmPC atual está disponível em www.roche.pl.
Interações
Os resultados dos estudos de metabolismo in vitro em microssomas hepáticos humanos indicam que o DM1 é metabolizado principalmente pela enzima CYP3A4 e em menor extensão pela CYP3A5. O uso concomitante de inibidores fortes do CYP3A4 (por exemplo, cetoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina e voriconazol) deve ser evitado, pois pode aumentar os níveis de DM1 e a toxicidade. Deve ser considerada uma formulação alternativa que não inibe ou apenas inibe ligeiramente CYP3A4. Se o uso concomitante de inibidores potentes do CYP3A4 não puder ser evitado e, sempre que possível, considere adiar a administração de trastuzumab emtansina até que os inibidores do CYP3A4 sejam eliminados de circulação (aproximadamente 3 semi-vidas dos inibidores). No entanto, se um inibidor forte do CYP3A4 for usado concomitantemente e o tratamento com trastuzumabe emtansina não puder ser adiado, o paciente deve ser monitorado de perto para efeitos adversos em tais casos.
A preparação contém a substância: Trastuzumab emtansina
Medicamento reembolsado: NÃO